

Clinical studies are essential for the development of new drugs, medical treatments, and therapies. Without them, it would be difficult to understand the efficacy and safety of drugs and different treatment modalities. However, the process of initiating a clinical study can be challenging and fraught with potential bottlenecks that can cause delays and increase costs. This blog discusses the key steps involved in initiating a clinical study and identifies the common bottlenecks that researchers and sponsors often encounter along the way.
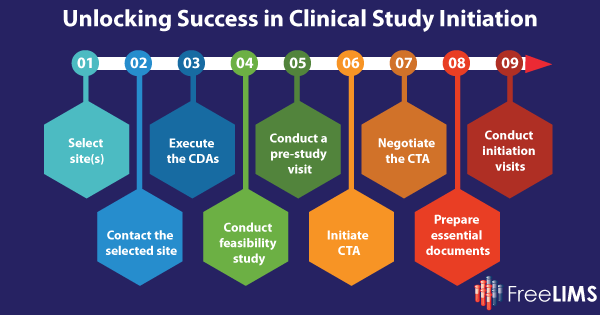
Step 1: Select site(s)
One of the first crucial steps in initiating a clinical study is selecting the appropriate sites where the research will take place. According to research from the Tufts Center for the Study of Drug Development (CSDD), site selection is a critical factor in the success of clinical trials. Shockingly, 37% of selected sites experience under-enrollment and 11% of sites fail to enroll a single subject. Poor enrollment can lead to substantial delays in the study timeline. Slow patient enrollment is cited as the top reason clinical trials fall behind schedule.
Step 2: Contact the Selected Sites
Once a list of investigative sites has been generated, the next step is to contact these sites and assess their interest in participating in the study. Communication with potential sites is typically handled via email, telephone, or fax, and responses are tracked using spreadsheets. While this may sound straightforward, it can quickly become cumbersome, especially when dealing with a large number of sites. Many sponsors rely on spreadsheets and email folders to track responses, but this approach can be inefficient. These are just a few bottlenecks that might be encountered at this stage.
Step 3: Execute the Confidentiality Disclosure Agreements (CDAs)
Sponsors must collect and fully execute Confidentiality Disclosure Agreements (CDAs) with all intended sites and investigators. Efficiency in this step can significantly impact the study’s timeline. The process of executing CDAs can vary based on the sponsor and standard operating procedures (SOPs) of the Contract Research Organization (CRO). Typically, the sponsor sends the CDA to individual sites via email, fax, or postal mail.
Step 4: Conduct Feasibility Study
Sponsors need to assess whether potential sites have the necessary facilities and knowledgeable staff to successfully enroll patients and produce high-quality data for the clinical trial. A standard practice is to use a Site Feasibility Questionnaire (SFQ) to assess sites’ interests and capabilities. A common bottleneck at this stage is poor accessibility and a lack of necessary facilities to support the study at potential sites.
Step 5: Conduct a Pre-Study Visit or Pre-Screening Visit
Pre-study visits (PSV) or phone calls are essential for sponsors to validate the information provided by potential sites, evaluate their capabilities, and review patient recruitment strategies. These visits can be time-consuming for sites and may not always be reimbursed, but they offer an opportunity to make a good impression and build a strong sponsor-site relationship.
Step 6: Initiate Clinical Trial Agreement (CTA)
The clinical trial agreement, also known as the contract, budget, or Investigator Commitment, is a critical business, legal, and financial element of the study startup process. Recent research indicates that contract cycle times have been doubling, leading to substantial delays in site activation. The CTA outlines responsibilities, obligations, and financial commitments between the sponsor and the trial site.
Step 7: Negotiate the CTA
Negotiating the CTA is a crucial step that often involves trading concessions based on the interests of both parties. Understanding each other’s perspectives and priorities is essential for successful and timely negotiations. Some sites may have internal policies that restrict their ability to negotiate, and these policies should be reviewed to avoid unnecessary barriers to participation in clinical trials.
Step 8: Prepare Essential Documents
Collecting regulatory documents, often referred to as “essential” documents, is vital for ensuring site eligibility and compliance. The guidance provided by the International Conference on Harmonization (ICH) on Good Clinical Practices (GCP) defines essential documents as those that, both individually and collectively, enable the assessment of the clinical trial’s conduct and the quality of the generated data. These documents play a crucial role in showcasing the adherence of the investigator, sponsor, and monitor to the standards of Good Clinical Practice, along with compliance with all relevant regulatory requirements. A lack of adequate skills in preparing these documents can be a bottleneck at this stage.
Step 9: Conduct Site Initiation Visits
The Site Initiation Visit (SIV) is the final step in the study startup process and involves training the Principal Investigator (PI) and their staff on the protocol and Good Clinical Practice (GCP) requirements. It is a mandatory step before any patient enrollment or protocol-specific activities can take place. Poor accessibility to the site and lack of adequate materials for staff training is a bottleneck to avoid.
Step 10: Wrap Up
While it might seem that study startup ends with the SIV, the reality is more complex. Initiating clinical studies is an ongoing process, and it’s not truly finished until the study meets its enrollment goals. Unfortunately, nearly 80% of clinical trials fail to meet these goals, with recruitment difficulties being a significant cause of study delays and failures.
Bottlenecks in Clinical Study
Bottlenecks are a pervasive issue in the initiation of clinical studies. These bottlenecks can manifest at various stages, from site selection and communication to contractual negotiations and regulatory document collection. Delays in any of these critical steps can significantly extend the study’s timeline, increase costs, and impede the timely development of new drugs and medical treatments. Identifying and addressing these bottlenecks is essential for sponsors and researchers to expedite the startup process, ensure that clinical trials proceed efficiently, and bring promising therapies to patients as quickly as possible.
Streamlining Clinical Study Initiation with a LIMS for Clinical Research & Trials
Laboratory Information Management Systems (LIMS) play a crucial role in overcoming bottlenecks during the initiation of clinical studies. A clinical LIMS can efficiently manage and organize the vast amounts of data and information involved in site selection, communication, regulatory compliance, and document management. A LIMS plays a key role in managing and streamlining the patient recruitment process. A LIMS for clinical research & trials can integrate with AI-based tools to screen and filter subject records that meet specific criteria for a particular study or research, simplifying the participant recruitment process. It also aids in associating research participants with the study in which they are enrolled.
A LIMS for clinical research & trials also enables sites to manage staff training and assess their competency to conduct participant enrollment and study-specific protocols or tasks. Moreover, a cloud-hosted LIMS enables sponsors, researchers, and site coordinators to access, track, and share critical information securely and seamlessly in real time. This not only accelerates the initiation process but also improves collaboration, ensures compliance with regulatory requirements, and ultimately aids in the successful execution of clinical trials.
Conclusion
Initiating a clinical study entails a number of progressive steps that must be executed meticulously if success is to be achieved. But along the way, researchers often encounter bottlenecks that may delay or derail the process. It is imperative for sponsors, researchers, and site coordinators to recognize and address these challenges early on to ensure the smooth and efficient startup of clinical trials. A LIMS for clinical research and trials has emerged as a valuable tool for overcoming these bottlenecks by streamlining data management, improving communication, and enhancing regulatory compliance.